法改正に伴う申請手続き
更新日:2024年10月9日
H26年に製造業は許可制・認定制から登録制に改められ、要件も簡素化されました。
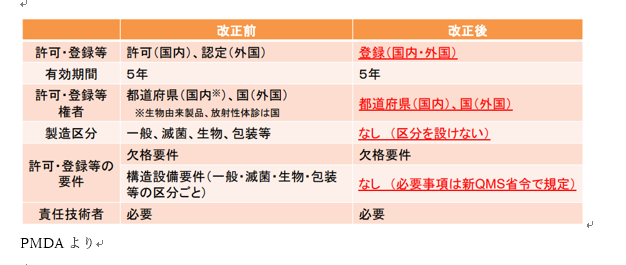
改正後は製造区分を設けず、登録すべき範囲も改められました。
①許可・登録等
製造業は登録に統一されました。
②有効期間
一度登録して終了ではなく、5年ごとに更新を行います。
有効期限の5か月前を目安に更新するようにしましょう。
③許可・登録等権者
国内の製造所なら都道府県、海外の製造所なら国(厚生労働大臣あて)に申請します。
④製造区分
区分は撤廃されました。
⑤許可・登録等の要件
申請者(法人であるときは、体外診断用医薬品の製造に関する業務を行う役員)は法第5条第3号イからへに該当しないこと(医薬品医療機器等法第23条の2の3の第4号)
イ.法第75条第1項の規定により許可を取り消され、取り消しの日から3年を経過していない者
ロ.法第75条の2第1項の規定により登録を取り消され、取り消しの日から3年を経過していない者
ハ.禁錮以上の刑に処せられ、その執行を終わり、又は執行を受けることがなくなった後、3年を経過していない者ニ.イからハまでに該当する者を除くほか、この法律、麻薬及び向精神薬取締法、毒物及び劇物取締法その他薬事に関する法令政令で定めるもの又はこれに基づく処分に違反し、その違反行為があった日から2年を経過していない者ホ.成年被後見人又は麻薬、大麻、あへん若しくは覚醒剤の中毒者
へ.精神の機能の障がいにより体外診断用医薬品製造業の業務を適正に行うにあたって必要な認知、判断及び意思疎通を適切に行うことができない者
⑥責任技術者(製造管理者)
医薬品医療機器等法第23条の2の14第5項により、薬剤師でなければならない。 各製造所ごとに置かなければならない。設計のみを行う製造所の管理者については、他の体外診断用医薬品の製造所の管理者との兼務が可能。ただし、設計のみを行う製造所と他の製造所における業務に支障を生じない範囲とすること。
体外診断用医薬品製造業の登録の範囲
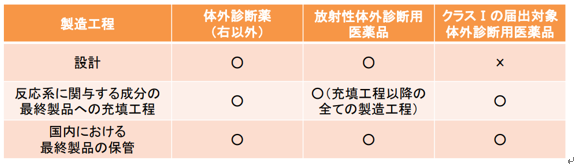
①設計
設計に関して最終的な責任を有する者の施設が登録対象となり(QMS調査が可能な施設)、
品目ごとに少なくとも1箇所の設計を行う製造所を特定。 クラスⅠ製品は承認又は認証不要であるため、
設計施設の登録は不要。
②反応系に関与する成分の最終容器への充填工程以降の全ての製造工程
放射性体外診断用医薬品の場合、「反応に関与する成分の最終容器への充填工程」から「国内における最終製品の保管」までの工程における全ての施設が登録対象となる。 それ以外の場合は、反応に関与する成分を直接の容器等へ充填する製造工程を行う施設が登録対象となります。
③国内における最終製品の保管
市場に製品を出荷するときの「出口」を対象とします。出荷判定を行う際に最終製品を保管している施設が登録対象。包装・表示のみを行っている施設は登録対象とはならない。
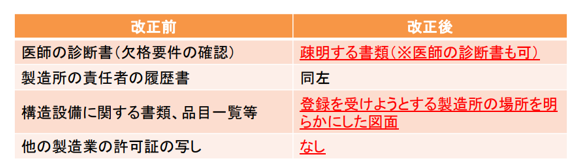
添付する資料
①国内製造所
(施行規則第114条の9)

②外国製造所
(施行規則第114条の15)
体外診断用医薬品に関する許認可のご相談はサポート行政書士法人へ
サポート行政書士法人では、新規で体外診断用医薬品業界へ参入される方から、既存の製造販売業者・製造業者・販売業者の皆さまに対して、医薬品医療機器等法に関する申請サポートやコンサルティングを行っています。
体外診断用医薬品の申請は専門性が高く、対応している行政書士が少ない分野の一つと言えます。
日々企業の皆様の代理人として行政庁への申請や折衝を行っている行政書士だからこそ蓄積できるノウハウ・実績を元に、体外診断用医薬品に関する法務サービスを提供いたします。
弊社の担当者は、全国の都道府県で申請実績があります。ぜひご相談ください。