法改正に伴う申請手続
更新日:2024年9月10日
目次
製造業の登録
新たに登録の対象となった工程・医療機器について
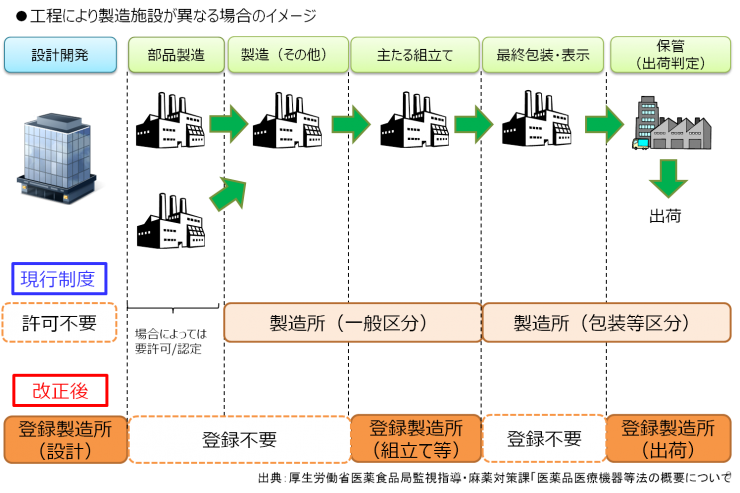
工程により製造施設が異なる場合の登録
既存の許可・認定製造所の取扱い
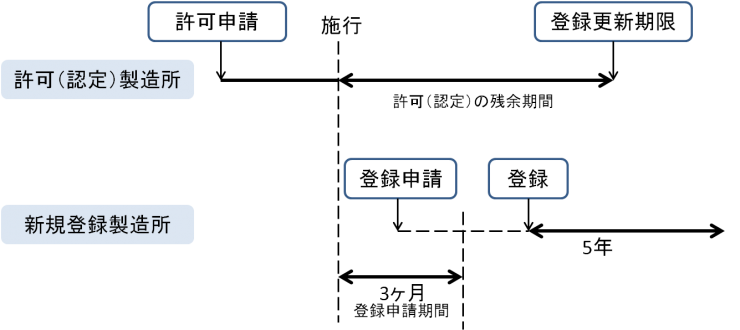
既存の許可・認定製造所のうち、登録対象になる製造所に該当するものは、登録を受けたものとみなされ、更新の期限についても、これまでの期限がそのまま登録の更新期限になります。
設計やプログラムなど、既存製品に関して新たに登録対象となる製造所については、改正法の施行日(平成26年11月25日)から起算して3ヶ月以内に登録申請を行う必要があります。
但し、該当施設が既に許可・認定を受けている製造所の場合は、登録製造所としてみなされ、新たな登録申請は必要ありません。

プログラムに関する製造販売業、製造業、販売業、品目の承認・認証
薬事法では、プログラムを使用している医療機器について、ハード部分に組み込んだ形で規制し、ソフト部分(プログラム)のみでは、規制の対象になりませんでしたが、法改正後は、ソフト部分のプログラム単体で、規制対象になります。
製造販売業については、他の医療機器同様に高度管理医療機器、管理医療機器の種類に応じて第一種又は第二種医療機器製造販売業許可を取得する必要があります。
プログラムが活用されている医療機器のクラス分類により、単体プログラムについても、品目の承認・認証が必要です。
医療機器プログラムについて電気通信回線を通じて提供を行う場合の業態は販売業として取り扱われます。
高度管理医療機器プログラムを電気通信回線を通じて提供しようとする場合は販売業の許可が、管理医療機器プログラムを提供しようとする場合は販売業の届出がそれぞれ必要となります。
添付文書の改訂
平成26年11月25日時点で、既に承認 、認証又は届出されている医療機器、11月25日時点で承認又は認証申請中の医療機器の添付文書については、平成29年11月25日までにできるだけ速やかに記載要領に基づいた改訂を行うことが必要です。
弊社は医療機器製造販売業者の皆さまに対して、医療機器等法施行に伴う添付文書の改訂・見直しについてのサポートメニューを提供しています。
添付文書の記載要領
| 医療機器等法施行後 | |
| 作成又は改訂年月 | 当該添付文書の作成又は改訂の年月及び版数を記載すること。改訂に当たっては、その履歴が分かるようにすることでその継続性を担保すること。 |
| 承認番号等 | 承認番号、認証番号又は届出番号のいずれかを記載するほか、単回使用の医療機器については、「再使用禁止」と記載すること。 |
| 類別及び一般的名称等 | 平成16年厚生労働省告示第298号により示される医療機器の一般的名称、JMDNコード、高度管理医療機器・管理医療機器・一般医療機器の別、特定保守管理医療機器・設置管理医療機器の別を記載すること。 なお、一つの承認、認証又は届出に係る医療機器に該当する一般的名称が複数になる場合、承認書、認証書又は届出書の一般的名称欄に記載した一般的名称等を記載するとともに、括弧書きで、承認書等の備考に記載されている一般的名称を記載すること。 |
| 販売名 | 販売名を記載すること。略称・愛称等、製品を特定する際に、使用者を混乱させるおそれがある名称は記載しないこと。 |
| 警告 | 当該医療機器の使用範囲内において、特に危険を伴う注意すべき事項を記載すること。「適用対象(患者)」、「併用医療機器」及び「使用方法」における警告事項についても小項目を作成し記載すること。 |
| 禁忌 ・禁止 | 当該医療機器の設計限界又は不適正使用等、責任範囲を超える対象及び使用方法を記載すること。「適用対象(患者)」、「併用医療機器」、及び「使用方法」における禁忌・禁止事項についても小項目を作成し記載すること。 |
| 形状・構造及び原理等 | 当該医療機器の全体的構造が容易に理解できるように、原則、イラスト図や写真、又はブロック図、原材料、構成品等を示すとともに、当該医療機器が機能を発揮する原理・メカニズムを簡略に記載すること。 |
| 使用目的又は効果 | 承認又は認証を受けた使用目的又は効果を記載すること。また、届出をした医療機器については、当該機器に係るクラス分類告示の一般的名称の定義の範囲内で記載すること。 |
| 品目仕様等 | 項目削除 |
| 使用方法等 | 設置方法、組立方法及び使用方法等について記載すること。なお、組み合わせて使用する医療機器がある場合は、その医療機器に対する要求事項又は組み合わせて使用可能な医療機器について記載すること。 |
| 使用上の注意 | 当該医療機器の使用に当たっての一般的な注意事項を記載すること。「適用対象(患者)」、「併用医療機器」及び「使用方法」等における注意事項についても小項目を作成し記載すること。 |
| 臨床成績 | 当承認、再審査又は使用成績評価申請時に用いられた臨床成績等を記載すること。 |
| 保管方法及び有効期間等 | 承認又は認証を受けた保管方法及び有効期間を記載すること。また、耐用期間又は使用期間を定めた医療機器においては、その根拠とともに記載すること。また、貯蔵・保管の条件や貯蔵・保管上の注意事項について記載すること。 |
| 取扱い上の注意 | 承認若しくは認証基準又は承認書、認証書若しくは届出書の中で取扱い上の注意事項が特に定められているものについては、その注意を記載すること。 |
| 保守・点検に係る事項 | 特定保守管理医療機器及び複数回使用する医療機器については、使用のために必要な保守・点検の項目やその点検頻度等について記載すること。複数回使用することが想定される医療機器については、洗浄、消毒、滅菌等の方法や手順について記載すること。 |
| 承認条件 | 承認条件が付された場合にその内容について記載すること。 |
| 包装 | 項目削除 |
| 主要文献及び文献請求先 | 文献請求先の氏名又は名称及び電話番号等を記載すること。 |
| 製造販売業者及び製造業者の氏名又は名称等 | 製造販売業者(選任製造販売業者を含む。)の氏名又は名称を記載すること。また、製造販売業者以外の製造業者が主たる設計を行う場合にあっては、当該製造業者の氏名又は名称を記載し、外国製造業者である場合はその国名、製造業者の英名を記載すること。 |
| 旧薬事法 | |
| 作成又は改訂年月 | 当該添付文書の作成又は改訂の年月日及び版数を記載すること。改訂に当たっては、 その履歴が分かるようにすることでその継続性を担保すること。 |
| 承認番号等 | 承認番号、認証番号又は届出番号のいずれかを記載する他、単回使用の医療機器につ いては、「再使用禁止」と記載すること。 |
| 類別及び一般的名称等 | 平成16年厚生労働省告示第298号により示される医療機器の一般的名称、JMDNコード、高度管理医療機器・管理医療機器・一般医療機器の別、特定保守管理医療機器・設置管理医療機器の別及び特定生物由来製品・生物由来製品の別を記載すること。なお、一つの承認、認証又は届出に係る医療機器に該当する一般的名称が複数になる場合、承認書、認証書又は届出書の一般的名称欄に記載した一般的名称等を記載するとともに、括弧書きで、承認書等の備考に記載されている一般的名称等を記載すること。 |
| 販売名 | 販売名を記載すること。略称・愛称等、製品を特定する際に、使用者を混乱させるお それがある名称は記載しないこと。特定生物由来製品及び生物由来製品であって、遺伝子組換え技術を応用して製造される場合にあっては、その旨を記載すること。 |
| 警告 | 当該医療機器の使用範囲内において、特に危険を伴う注意すべき事項を記載すること。「適用対象(患者)」、「併用医療機器」及び「使用方法」における警告事項についても小項目を作成し記載すること。 |
| 禁忌 ・禁止 | 当該医療機器の設計限界又は不適正使用等、責任範囲を超える対象及び使用方法を記載すること。「適用対象(患者)」、「併用医療機器」及び「使用方法」における禁忌・禁止事項についても小項目を作成し記載すること。 |
| 形状・構造及び原理等 | 当該医療機器の全体的構造が容易に理解できるように、ブロック図、組成式又は構成 金属組成等により概略を記載すること。また、当該医療機器が機能を発揮する原理・メカニズムを簡略に記載すること。 |
| 使用目的、効能又は効果 | 承認を受けた使用目的、効能又は効果を記載すること。認証を受けた医療機器については、認証を受けた使用目的、効能又は効果を記載すること。届出をした医療機器については、当該機器に係るクラス分類告示の一般的名称の定義を記載すること(平成16年7月20日付け医薬食品局長通知薬食発第0720022号参照)。 |
| 品目仕様等 | 承認書、認証書又は届出書において、品目仕様欄に記載した項目のうち性能に関する事項について簡潔に記載すること。 |
| 操作方法又は使用方法等 | 設置方法、組立方法及び使用方法等について記載すること。なお、組み合わせて使用する医療機器がある場合は、その医療機器に対する要求事項若しくは組み合わせて使用可能な医療機器について記載すること。 |
| 使用上の注意 | 当該医療機器の使用に当たっての一般的な注意事項を記載すること。「適用対象(患者)」、「併用医療機器」及び「使用方法」における注意事項についても小項目を作成し記載すること。 |
| 臨床成績 | 承認申請時に用いられた臨床成績又は製造販売後臨床試験の結果等を記載すること |
| 貯蔵・保管方法及び使用期間等 | 貯蔵・保管方法、(一回使用あたりの)使用期間、有効期間・使用の期限(耐用期間の設定が必要な医療機器)を小項目を設けて記載すること。 |
| 取扱い上の注意 | 基準又は承認書、認証書若しくは届出書の中で取扱い上の注意事項が特に定められているものについては、その注意を記載すること。 |
| 保守・点検に係る事項 | 特定保守管理医療機器及び複数回使用する医療機器については、再使用のために必要な措置(滅菌、維持・管理、保守・点検等)を記載すること。 |
| 承認条件 | 承認条件が付された場合に記載すること。 |
| 包装 | 包装単位を記載すること。 |
| 主要文献及び文献請求先 | 文献請求先の氏名又は名称及び住所を記載すること。 |
| 製造販売業者及び製造業者の氏名又は名称及び住所等 | 製造販売業者の氏名又は名称、住所及び電話番号を記載すること。また、製造業者の氏名又は名称を記載し、外国製造所で製造される医療機器の場合にあっては外国製造所の国名、製造業者の英名を記載すること。 |
引用元:「医療機器の添付文書の記載要領について」(平成17年3月10日付け薬食発0310003号)・「医療機器の添付文書の記載要領の改正について」(平成26年10月2日付け薬食発1002第8号)
既存医療機器の承認、認証、届出の記載整備
平成26年11月25日時点で、既に承認 、認証又は届出されている医療機器については、承認申請書等の製造方法欄及び製造販売する品目の製造所欄が、医療機器等法により変更となったことから、記載内容の整備を行う必要があります。
弊社は医療機器製造販売業者の皆さまに対して、医療機器等法施行に伴う記載整備についてのサポートメニューを提供しています。
平成26年11月医療機器等法施行に伴う記載整備の方法・期限・提出先
記載整備の方法
記載整備の方法は、軽微変更届出の様式にて製造方法欄と製造販売する品目の製造所欄の記載を整備を行うことになります。
様式63 の10(1):医療機器製造販売承認事項軽微変更届書
様式63 の24(1):外国製造医療機器製造販売承認事項軽微変更届書
様式66(1):指定高度管理医療機器・指定管理医療機器認証事項軽微変更届書
様式66(3):外国製造指定高度管理医療機器・指定管理医療機器製造販売認証事項軽微変更届書
様式40:医薬品・体外診断用医薬品・医薬部外品・化粧品・医療機器製造販売届出事項変更届書
記載整備の届出先
届出先は、承認品目・届出品目は独立行政法人医薬品医療機器総合機構理事長、認証品目は認証を受けた登録認証機関宛てとし、品目ごとに届け出る必要があります。
記載整備の期限
期限としては、改正法施行時において、製造販売業者が保有する品目のうち、新法第23 条の2の5第6項又は第23 条の2の23 第3項に基づく承認又は認証の取得後、5年ごとに受けるべきQMS調査について、当該期間の残存期間が最も長い品目のQMS調査を受けるべき日から30 日後までに、当該製造販売業者が保有する全ての承認品目又は認証品目の記載整備を完了させることとなっています。
記載整備期限前であっても、新法に基づくQMS調査を行った品目については調査後に当該品目を含めた関連品目の記載整備を行うなど、可能なものは速やかに記載整備することが望ましいとされています。
また、記載整備期限の前に製造方法欄又は製造販売する品目の製造所欄の承認(認証)事項を一部変更承認(認証)申請又は軽微変更届出により変更する場合は、変更の機会に併せて該当する欄を記載整備する必要があります。
クラスⅠの医療機器に関しては、改正法施行前に製造販売届書により届出された品目に関しては、製造方法欄又は製造販売する品目の製造所欄に変更が生じない限り、新法に対応した記載整備は必要ないが、当該欄の変更が生じた際には、その変更にあわせて記載を整備することが必要です。
医療機器に関する許認可のご相談はサポート行政書士法人へ
サポート行政書士法人では、新規で医療機器業界へ参入される方から、既存の製造販売業者・製造業者・販売業者の皆さまに対して、医薬品医療機器等法に関する申請サポートやコンサルティングを行っています。
医療機器の申請は専門性が高く、対応している行政書士が少ない分野の一つと言えます。
日々企業の皆様の代理人として行政庁への申請や折衝を行っている行政書士だからこそ蓄積できるノウハウ・実績を元に、医療機器に関する法務サービスを提供いたします。
弊社の担当者は、全国の都道府県で申請実績があります。ぜひご相談ください。

専任スタッフが全国の案件を対応しています。
