更新日:2024年3月20日
制造业的注册
关于新的需要注册的工程和医疗器械
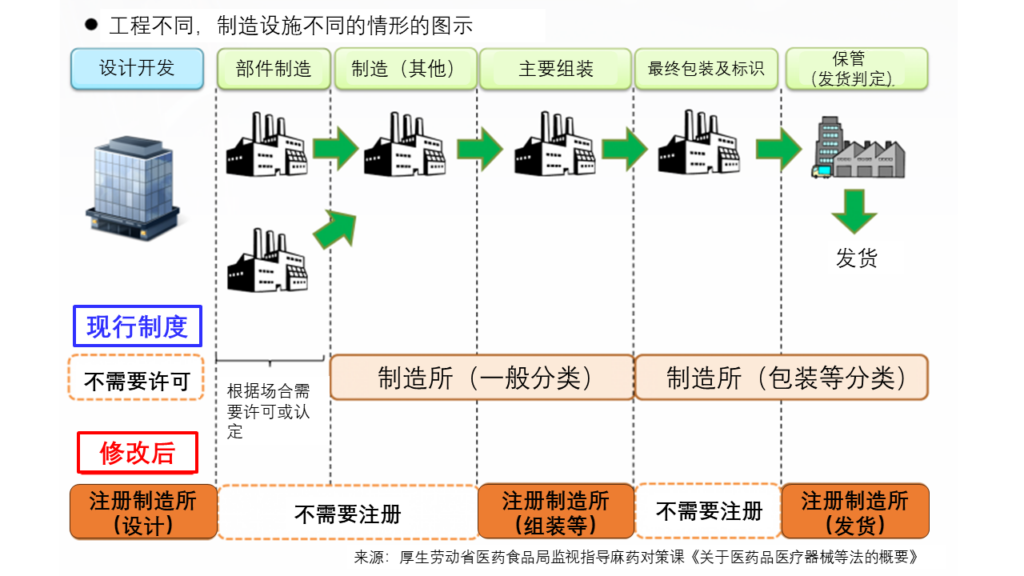
工程不同而制造设施不同的情形的注册
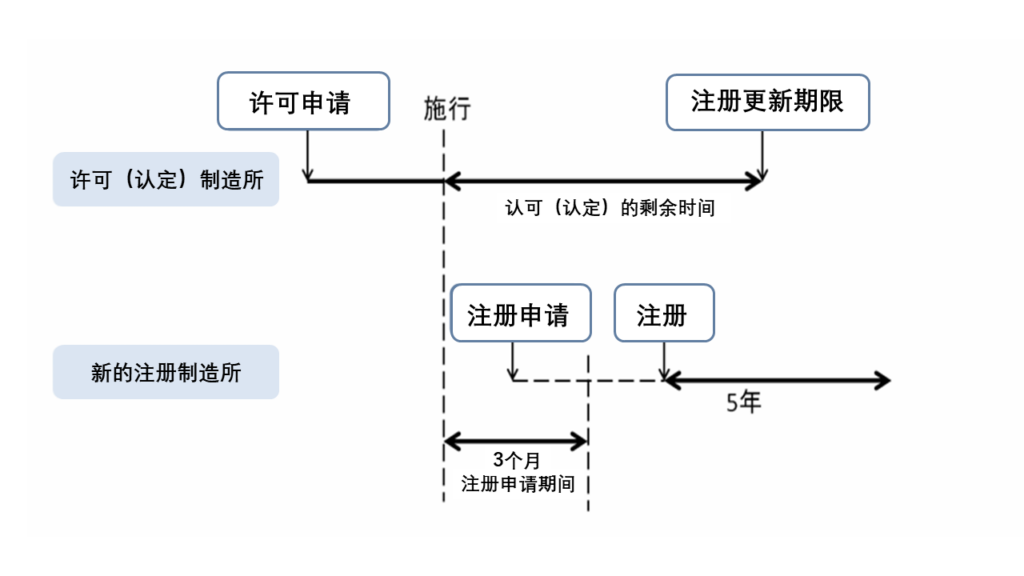
现有的许可或认定制造所的处理
现有的许可或认定的制造所中,变为需要进行注册的制造所的,被视为已经进行注册,注册的更新日期与之前的期限相同。
与设计和软件等现有产品相关的新的需要进行注册的制造所,需在法律修改施行日(平成26年11月25日)之后的3个月内提交注册申请。
但是,如果相关设施是已经获得许可和认证的制造所,则被视为是已经注册的制造所,无需进行新的注册申请。
软件相关制造贩卖业、制造业、贩卖业、品目的承认和认证
根据药事法,对于使用软件的医疗设备,只有将软件嵌入硬件部分时才受到规范,软件部分(软件)本身不是受到规范的对象,然而在法律修改后,单独的软件本身也成为受到规范的对象。
对于制造贩卖业,与其他医疗设备一样,需根据高度管理医疗器械、管理医疗器械的种类取得第一种或第二种医疗器械制造贩卖业许可。
根据使用该软件的医疗器械的级别分类,单独的软件也需要获得品目的承认或认可。
通过网络提供医疗器械软件的业务被视为贩卖业。
如果想通过电信线路提供高度管理医疗器械软件,则需要取得贩卖业的许可,如果想提供管理医疗器械软件,则需要取得贩卖业的备案。
对附件文书修改
对于截至平成26年11月25日已经获得承认、认证或备案的医疗器械,以及截至11月25日正在申请承认或认证的医疗器械,必须在平成29年11月25日前尽快按照说明指南修订附件文书。
我们为医疗器械制造销售企业提供有关医疗器械等法实施而导致的附件文书修订和改正相关的支持服务。
附件文书的记载要领
| 医药器械等法施行后 | | 编制或修订日期 | 应在该附件文书中记载编制或修订的年月和版本号。修订时,应提供清晰的修订历史记录,以确保其连续性。 | | 承认号码等 | 除记载承认号码、认证号码或备案号码外,对于一次性使用的医疗器械,还应标明”禁止再次使用”的字样。 | | 类别及一般名称等 | 需记载根据厚生劳动省公告第298号指定的医疗器械的一般名称、JMDN代码,以及其分类(高度管理医疗器械、管理医疗器械、一般医疗器械;特定保养管理医疗器械、设置管理医疗器械)。另外,如果适用于承认、认证或者备案的医疗器械的一般名称有多个的情况下,应该记载在承认书、认证书或者备案书的一般名称栏中记载的一般名称,并用括号注明在承认书等的备注栏中记载的一般名称。 | | 贩卖名称 | 应记载贩卖名称。不要记载简称、昵称等会导致使用者在识别制品时产生混淆的名称。 | | 警告 | 应该记载在该医疗器械使用范围内特别需要注意的危险事项。在“适用对象(患者)”、“联合使用医疗器械”和“使用方法”方面的警告事项,应创建小标题进行详细记录。 | | 禁忌 ・禁止 | 应记载该医疗器械的设计限制或不当使用等超出责任范围的对象和使用方法。在“适用对象(患者)”、“联合使用医疗器械”和“使用方法”方面的禁忌或禁止事项,应创建小标题进行详细记录。 | | 形状或构造及原理等 | 为了便于理解该医疗器械的整体构造,原则上,应展示插图、照片或框图、原材料、部件等,并简略地记载该器械发挥功能的原理和机制。 | | 使用目的或效果 | 应记载已获得承认或认证的使用目的或效果。并且,已经备案的医疗器械,应在该器械所适用的级别分类公告的一般名称的定义的范围进行记载。 | | 品目规格等 | 项目删除 | | 使用方法等 | 应记载设置方法、组装方法和使用方法。如果组合使用的医疗器械的情况下,应记载对该医疗器械的要求事项或者能组合使用的医疗器械。 | | 使用上的注意 | 应记载使用该医疗设备时的一般注意事项。在“适用对象(患者)”、“联合使用医疗器械”和“使用方法”方面的注意事项,应创建小标题进行详细记录。 | | 临床成绩 | 应记载用于承认、再次审查或使用成绩评价申请的临床成绩等。 | | 保管方法及有效期间等 | 应记载获得承认或认证的保管方法和有效期间。并且,对于有耐用期限或使用期限的医疗设备,应记载该期限及依据。此外,应记载贮藏或保管的条件和贮藏或保管上的注意事项。 | | 使用上的注意 | 承认或认证基准或承认书、认证书或备案书中特别规定了使用上的注意事项,应记载该注意。 | | 保养或点检相关事项 | 对于特定保养管理医疗器械和多次使用的医疗器械,应记载为了使用所需的维护和点检项目及检查频率等。对于预计将被多次使用的医疗器械,应记载清洁、消毒和灭菌的方法和步骤。 | | 承认条件 | 如果存在附加的承认条件应记载这些内容。 | | 包装 | 项目削除 | | 主要文献及文献获取对象 | 应记载文献获取对象的名字或名称以及电话等 | | 制造贩卖业者以及制造业者的名字或名称等 | 应记载制造贩卖业者(包含选任制造贩卖业者)的姓名或名称。当由制造贩卖业者以外的制造业者进行主要设计时,应记载该制造业者的姓名或名称,如果是外国制造业者,则需要记载其国家名称和制造业者的英文名称。 |
| | | 旧药事法 | | 编制或修订日期 | 应在该附件文书中记载编制或修订的年月和版本号。修订时,应提供清晰的修订历史记录,以确保其连续性。 | | 承认号码等 | 除记载承认号码、认证号码或备案号码外,对于一次性使用的医疗器械,还应标明”禁止再次使用”的字样。 | | 类别及一般名称等 | 需记载根据厚生劳动省公告第298号指定的医疗器械的一般名称、JMDN代码,以及其分类(高度管理医疗器械、管理医疗器械、一般医疗器械;特定保养管理医疗器械、设置管理医疗器械;特定生物由来制品、生物由来制品)。另外,如果适用于承认、认证或者备案的医疗器械的一般名称有多个的情况下,应该记载在承认书、认证书或者备案书的一般名称栏中记载的一般名称,并用括号注明在承认书等的备注栏中记载的一般名称。 | | 贩卖名称 | 应记载贩卖名称。不要记载简称、昵称等会导致使用者在识别制品时产生混淆的名称。如果是特定生物由来制品和应用转基因技术的生物由来制品,应该记载这一情况。 | | 警告 | 应该记载在该医疗器械使用范围内特别需要注意的危险事项。在“适用对象(患者)”、“联合使用医疗器械”和“使用方法”方面的警告事项,应创建小标题进行详细记录。 | | 禁忌 ・禁止 | 应记载该医疗器械的设计限制或不当使用等超出责任范围的对象和使用方法。在“适用对象(患者)”、“联合使用医疗器械”和“使用方法”方面的禁忌或禁止事项,应创建小标题进行详细记录。 | | 形状或构造及原理等 | 为了便于理解该医疗器械的整体构造,应通过框图、组成公式或组成金属成分等进行概要记载。并且,应简略地记载该器械发挥功能的原理和机制。 | | 使用目的或效果 | 应记载已获得承认的使用目的或效果。已经获得认证的医疗器械,应记载已获得认证的使用目的,效能或效果。已经备案的医疗器械,应记载该器械所适用的级别分类公告的一般名称的定义(参照平成16年7月20日医薬食品局长通知药食发第0720022号)。 | | 品目规格等 | 应简洁地记载在承认书、认证书或备案书的品目规格栏中记载的项目中与性能相关的事项。 | | 操作方法或使用方法等 | 应记载设置方法、组装方法和使用方法。如果组合使用的医疗器械的情况下,应记载对该医疗器械的要求事项或者能组合使用的医疗器械。 | | 使用上的注意 | 应记载使用该医疗设备时的一般注意事项。在“适用对象(患者)”、“联合使用医疗器械”和“使用方法”方面的注意事项,应创建小标题进行详细记录。 | | 临床成绩 | 应记载用于承认申请的临床结果或制造贩卖后临床试验的结果等。 | | 贮藏或保管方法及使用期间等 | 应通过创建小标题的形式,记载贮藏或保管方法,(每次使用的)使用期间、有效期间或使用的期限(需要设定耐用期限的医疗器械)。 | | 使用上的注意 | 基准或承认书、认证书或备案书中特别规定了使用上的注意事项,应记载该注意。 | | 保养或点检相关事项 | 对于特定保养管理医疗器械和多次使用的医疗器械,应记载为了再次使用所需采取的必要措施(灭菌、维护或管理、保养或点检等) | | 承认条件 | 应记载存在的附加承认条件。 | | 包装 | 应说明包装单位 | | 主要文献及文献获取对象 | 应记载文献获取对象的名字或名称以及住所等 | | 制造贩卖业者以及制造业者的名字或名称以及住所等 | 应记载制造贩卖业者(包含选任制造贩卖业者)的姓名或名称、住所以及电话号码。并且,应记载制造业者的姓名或名称,如果是外国制造所制造的医疗器械,应记载外国制造所的国家名称、制造业者的英文名称。 |
|
引用来源:《关于医疗器械的附件文书的记载要领》 “(平成17年3月10日 药食发0310003号)和《关于医疗器械的附件文书的记载要领的改正》(2014年10月2日 药食发1002第8号)
既存医療機器の承認、認証、届出の記載整備
对于截至2014年11月25日已经获得承认、认证或备案的医疗器械,因为根据医疗器械等法,承认申请书等的制造方法栏和制造贩卖产品的制造所栏发生了变更,所以需要调整相应的记载内容。
我们为医疗器械制造贩卖企业提供与因医疗器械法等的实施而引起的记载调整相关的一系列服务。
平成26年11月医疗器械等法实施所导致的记载调整的方法、期限和提出对象
记载调整的方法
记载的调整方法是通过轻微变更备案的样式,对制造方法栏和制造贩卖的产品的制造所栏的记载进行调整。
样式63之10(1): 医疗器械制造贩卖承认事项轻微变更备案书
样式63之24(1): 外国制造医疗器械制造贩卖承认事项轻微变更备案书
样式66(1):指定高度管理医疗器械或指定管理医疗器械承认事项轻微变更备案书
样式66(3):外国制造指定高度管理医疗器械或指定管理医疗器械承认事项轻微变更备案书
样式40:医药品/体外诊断用医药品/非医药品/化妆品/医疗器械制造贩卖备案事项变更备案书
记载调整的备案提出对象
备案提出对象为,承认品目和备案品目为独立行政法人医药品医疗器械综合机构的理事长,认证品目为获得认证的注册认证机构,每个品目都需要单独备案。
记载调整的期限
在修正的法律实施时,制造贩卖业者保有的品目中,基于新法第23条之2之5第6款或者23条之2之23第3款取得承认或认证后,每五年应接受QMS调查,该制造贩卖业者应在该期间残存期间最长的品目的应进行QMS调查日起30日内,完成其所保有的全部承认品目或认证品目的记载调整。
即使在记载调整期限之前,对于已进行新法下的QMS调查的项目,建议尽快进行相关品目的记载调整工作,包括在调查后对该项目及相关品目进行记载调整。
另外,如果在记载调整期限之前,通过部分变更承认(认证)申请或轻微变更备案对制造方法栏或制造贩卖品目的制造所栏的承认(认证)事项进行变更时,那么需要在变更时一并完成相应栏目的记载调整工作。
对于I类医疗器械,在修正的法律施行前通过制造贩卖备案书进行备案的品目,只要制造方法栏或制造贩卖品目的制造所栏没有发生变更,就不需要进行与新法相符的记载调整。但是,如果相关栏目发生变更,就需要根据变更进行相应的记载调整。
咨询有关医疗器械的许可、认可及承认等相关事宜,请联系我们。
我们向新进入医疗器械行业的企业,以及现有的制造商、生产商和经销商提供与医药品医疗器械等法相关的申请支持和咨询服务。
医疗器械的申请具有高度专业性,能够完美处理这些业务的行政书士很少。
而我们提供医疗器械相关的法律服务的基础是我们作为企业代理人向日本行政机关申请与斡旋沟通时所积累的丰富经验和优秀业绩。
我们的团队拥有在全国各都道府县的申请经验。请务必与我们联系以获取更多信息。
专任人员处理全日本的案件。